
Aspen/HYSYS/ProII如何输入自定义组分
本文最后更新于 2024-10-18,文章内容距离上一次更新已经过去了很久啦,可能已经过时了,请谨慎参考喵。
title: Aspen/HYSYS/ProII如何输入自定义组分
top_img: false
tags:
- Aspen
- HYSYS
- ProII
categories:
- 化工
cover: '/upload/cdn4files/202306151013455.png'
abbrlink: 78dc3dab
date: 2023-06-15 09:43:46
copyright:
comments:
前言
通常在工艺设计中我们会使用到aspen/hysys/proii等稳态模拟软件来计算一些工艺参数
对于已知的体系,数据库中现有的物质组分,可以很好的输入相关的参数
但是总会遇到一些数据库中没有的组分,这个时候就需要自己去输入相关的参数来估算其性质
本文就以aspen/hysys/proii为例,简单说明一下如何输入自定义组分
注意:为了方便理解,本文三个软件中都以对氯甲苯举例,实际按照自己需要的组分进行设置即可
Aspen
新建aspen文件,来到组分输入界面:
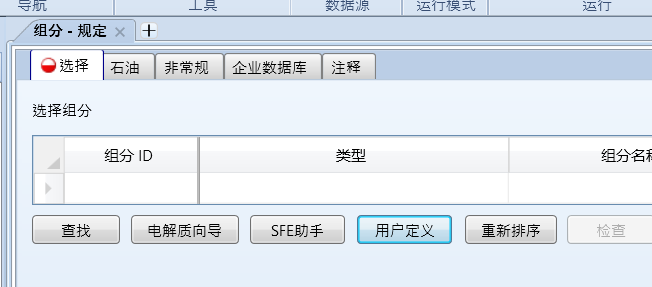
打开用户定义:
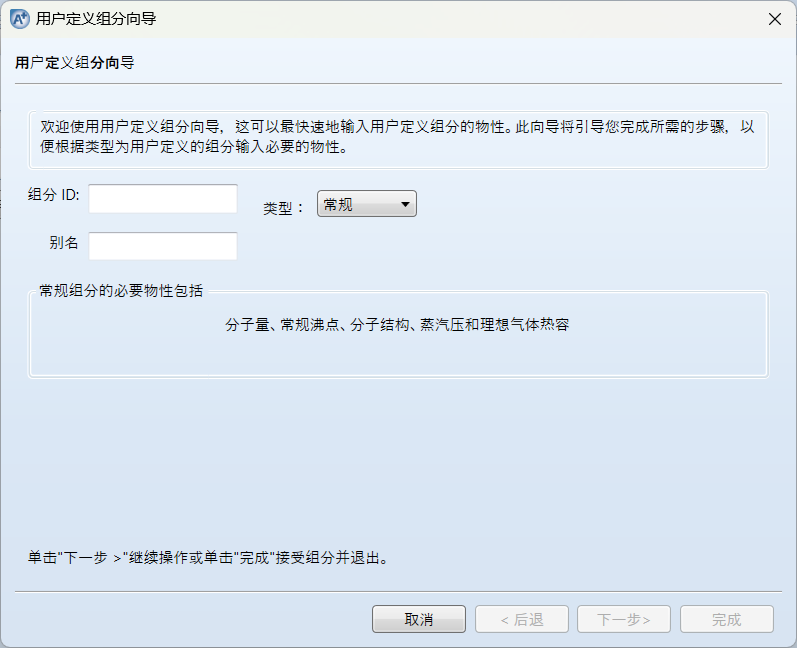
输入组分名称DLJB后点击下一步:
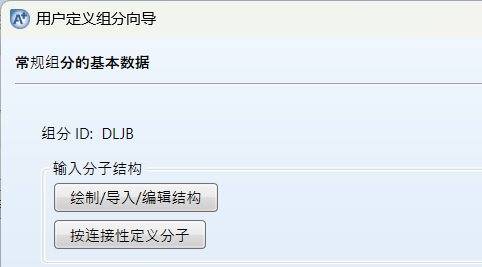
点击绘制结构:
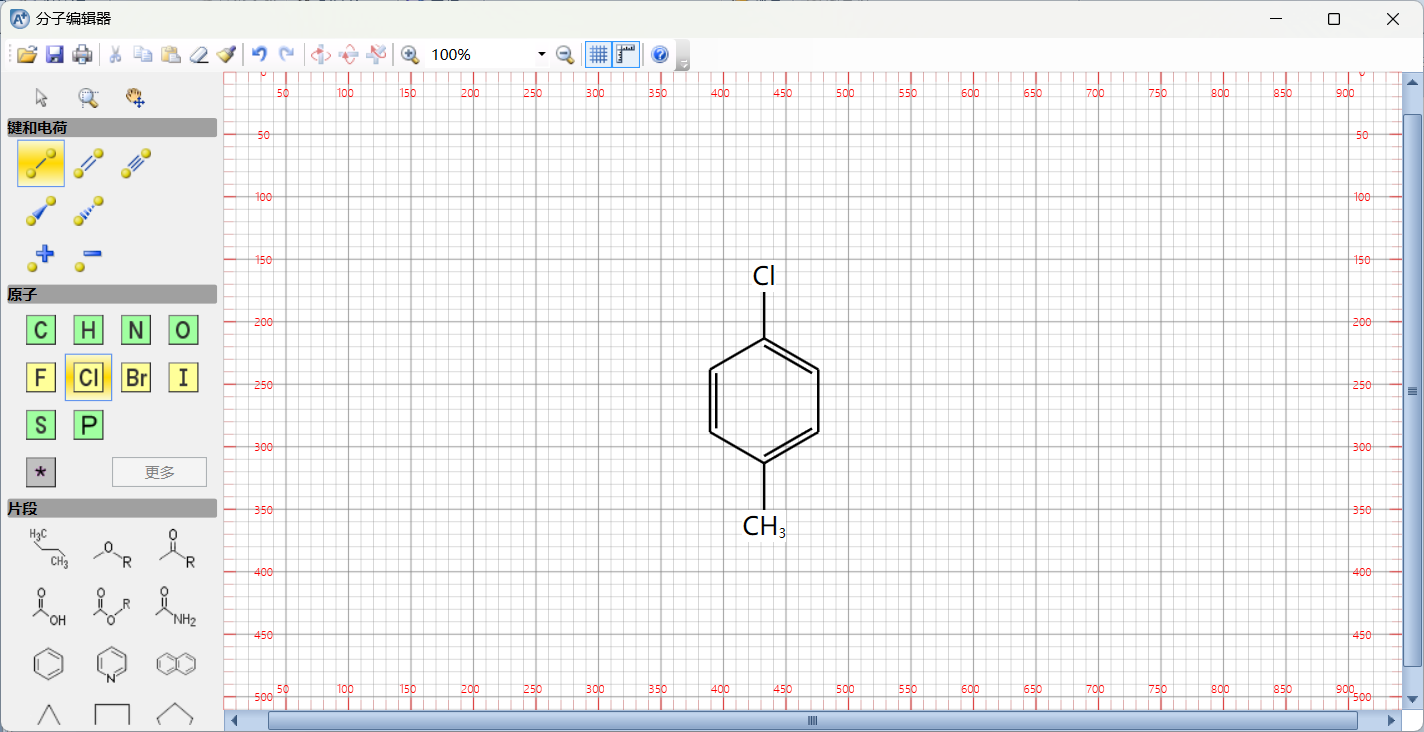
绘制好结构之后直接点击右上角关闭即可,如果需要保存这个分子结构也可以点击左上角另存为一个分子结构的文件
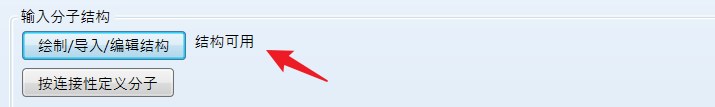
此时会显示结构可用
继续输入参数:
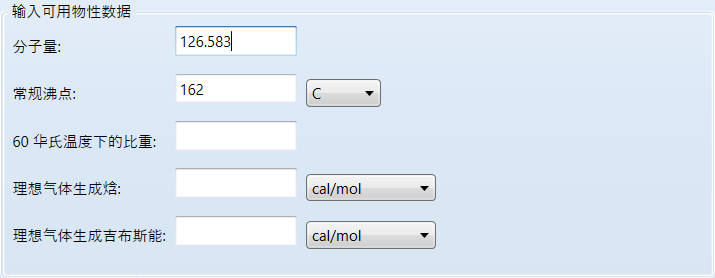
aspen要求至少输入分子量和常规沸点这两个参数,当然其他参数如果也有输入进去会估算的更准确
输入完毕之后点击下一步:
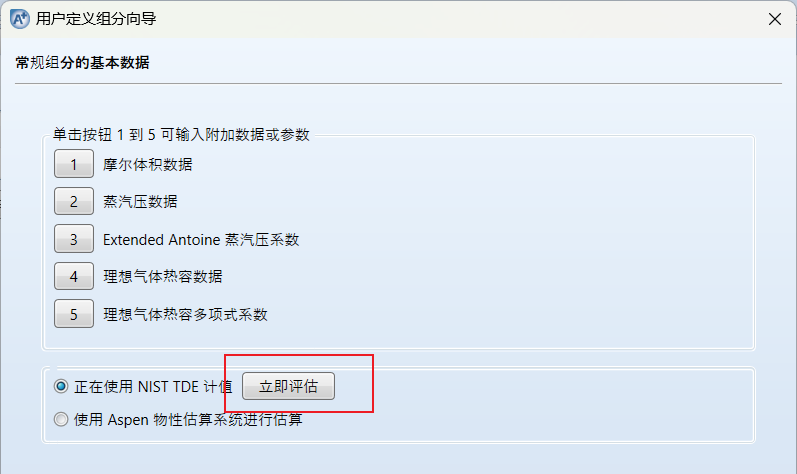
上面的1-5参数也是同理,如果有,点击输入,如果没有,不用输入也可以
输入完成之后点击立即评估,点击确定
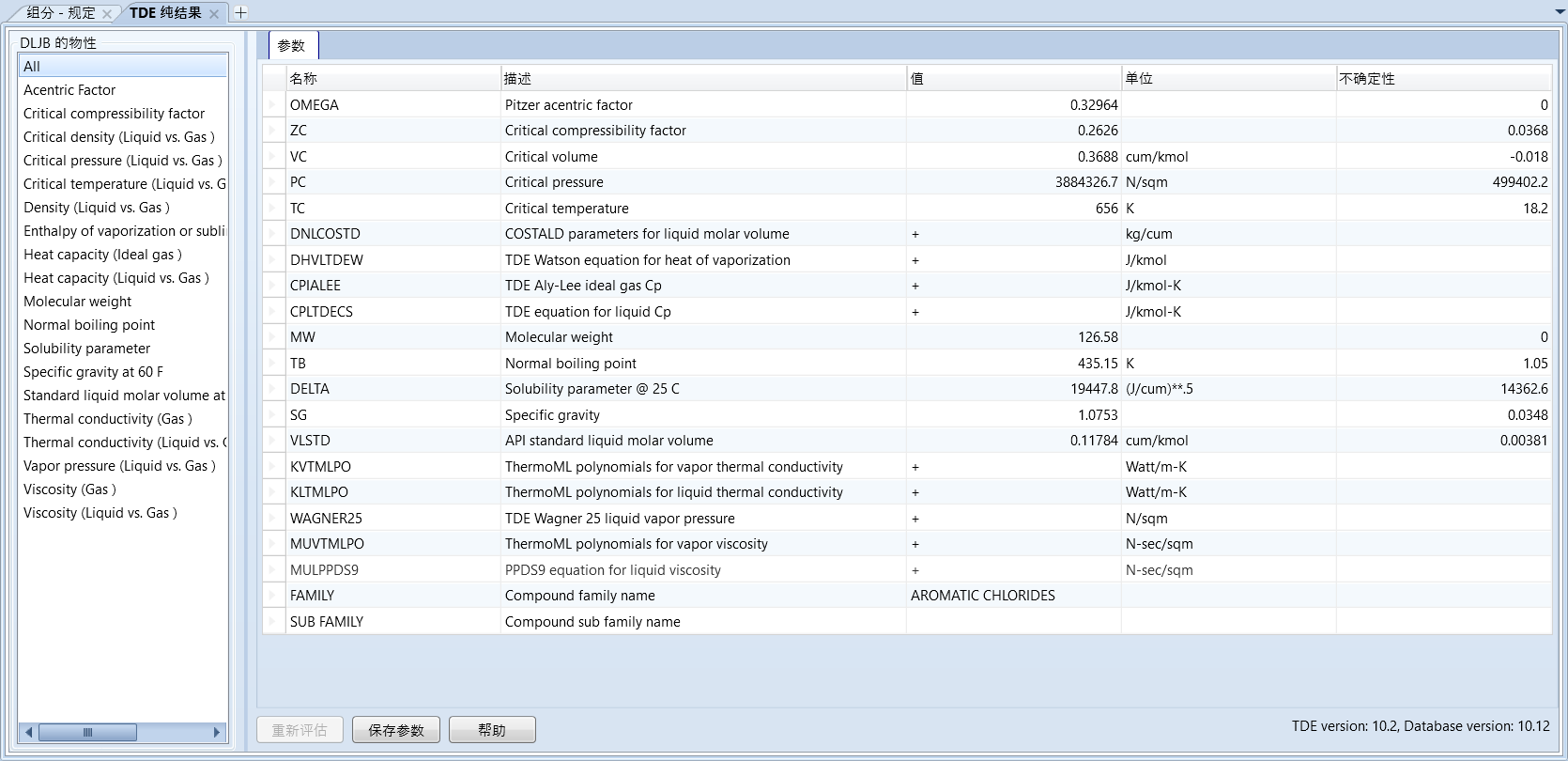
此时aspen会估算出大致的物性并显示出来,点击保存参数,确定即可输入完毕
如果物性不全,或者偏差较大,可以继续使用UNIFAC基团贡献法继续估算
打开组分-分子结构:
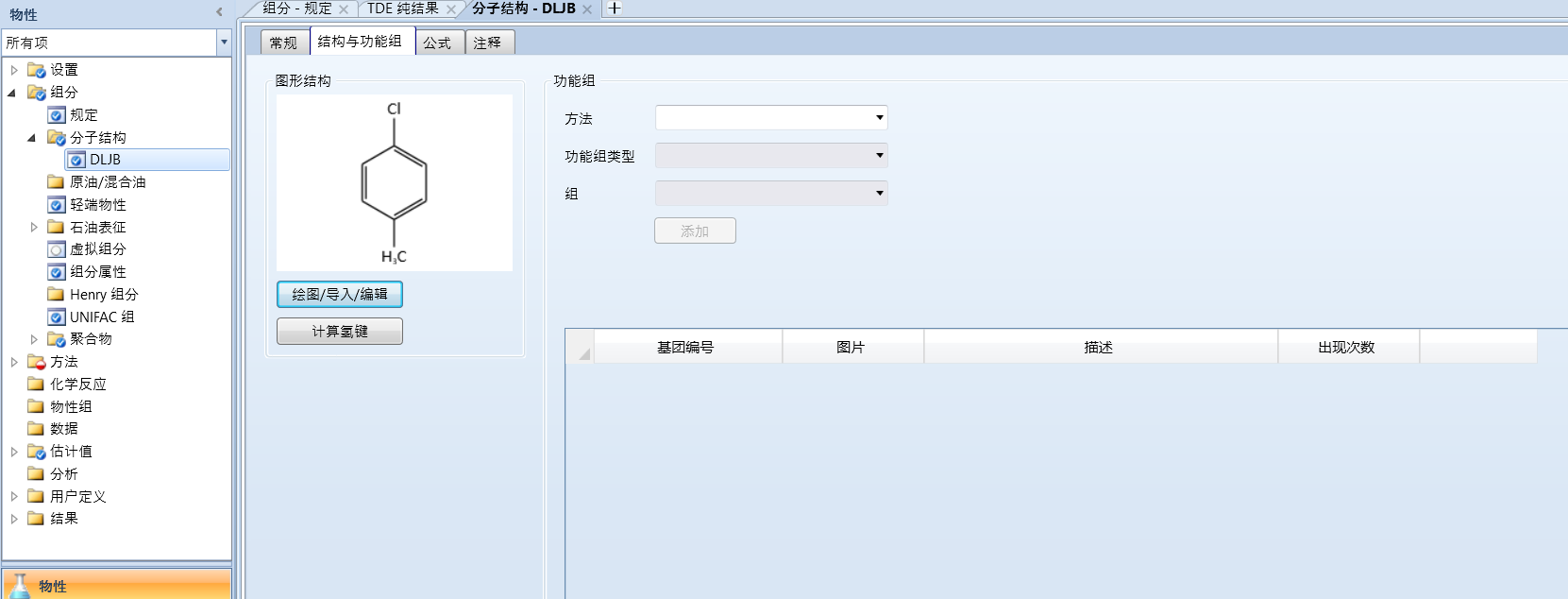
右侧功能组的方法选择UNIFAC
选择分子结构中的基团进行添加,如图所示:
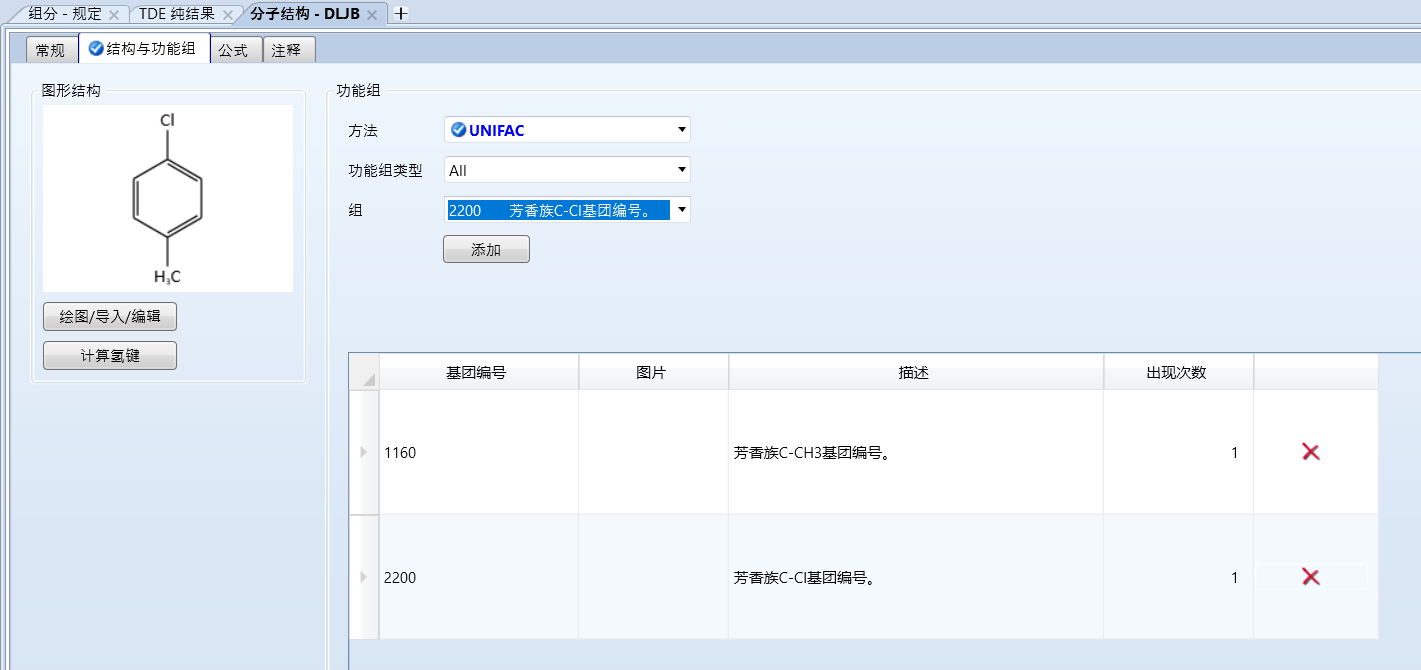
aspen中好像不用单独输入苯环基团
对于其他分子结构的组分也是同理,输入物质中含有的基团和数量即可
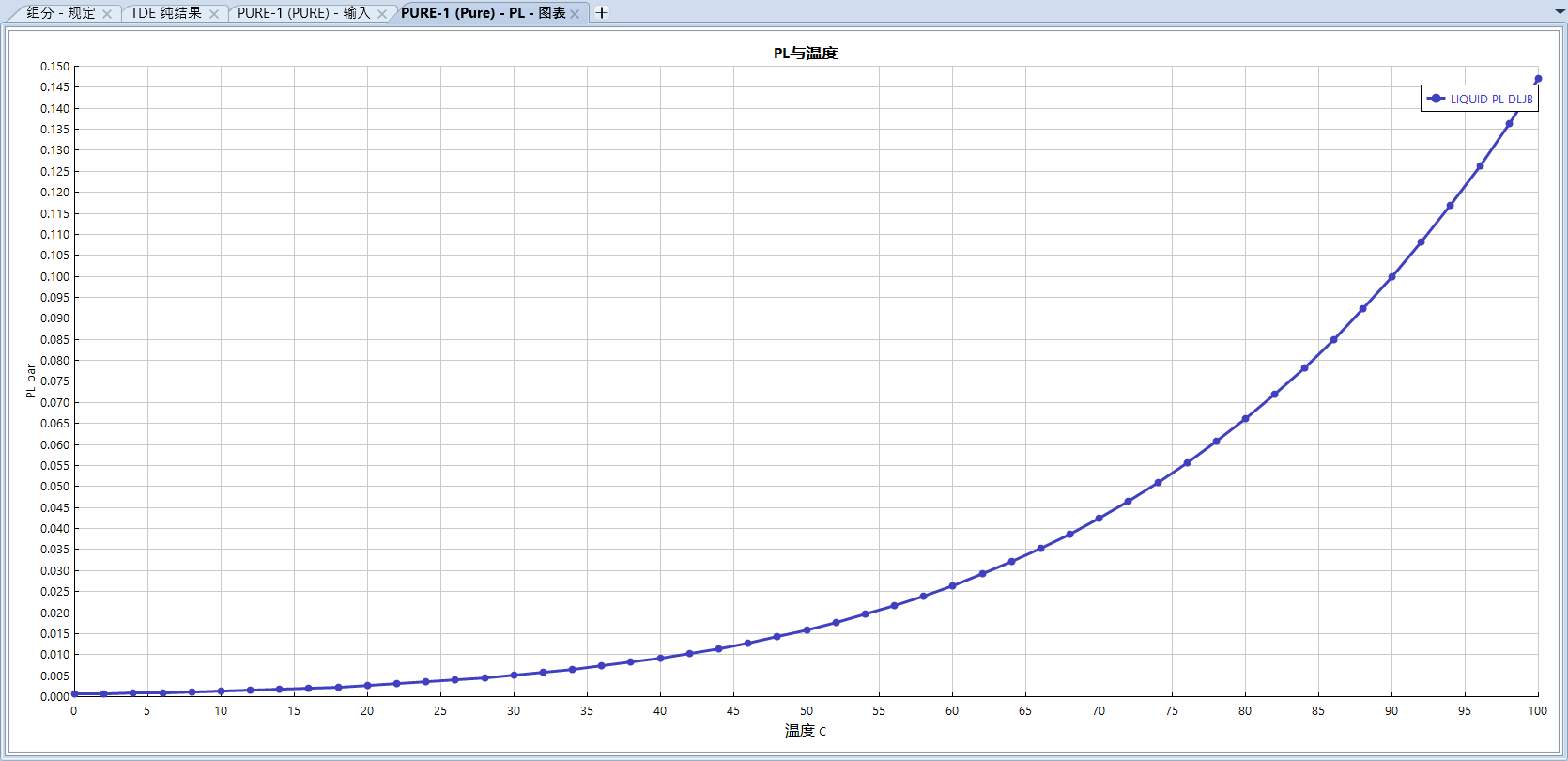
选择好热力学方法之后点击运行,即可估算出相对应的物质参数,完成模拟
例如PL饱和蒸汽压:
HYSYS
新建文件:
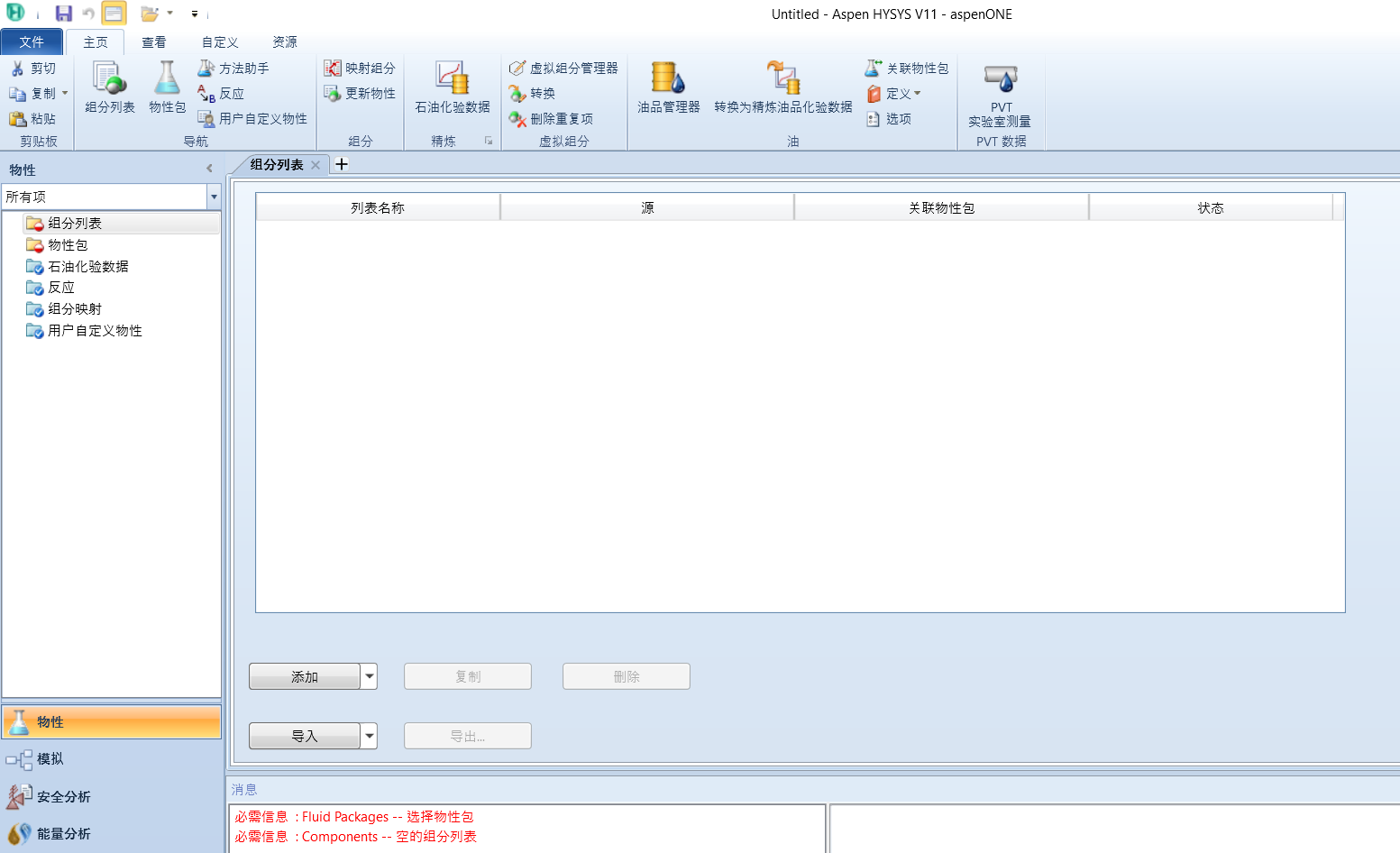
点击添加,左上角点击虚拟组分,点击创建或编辑虚拟组分:
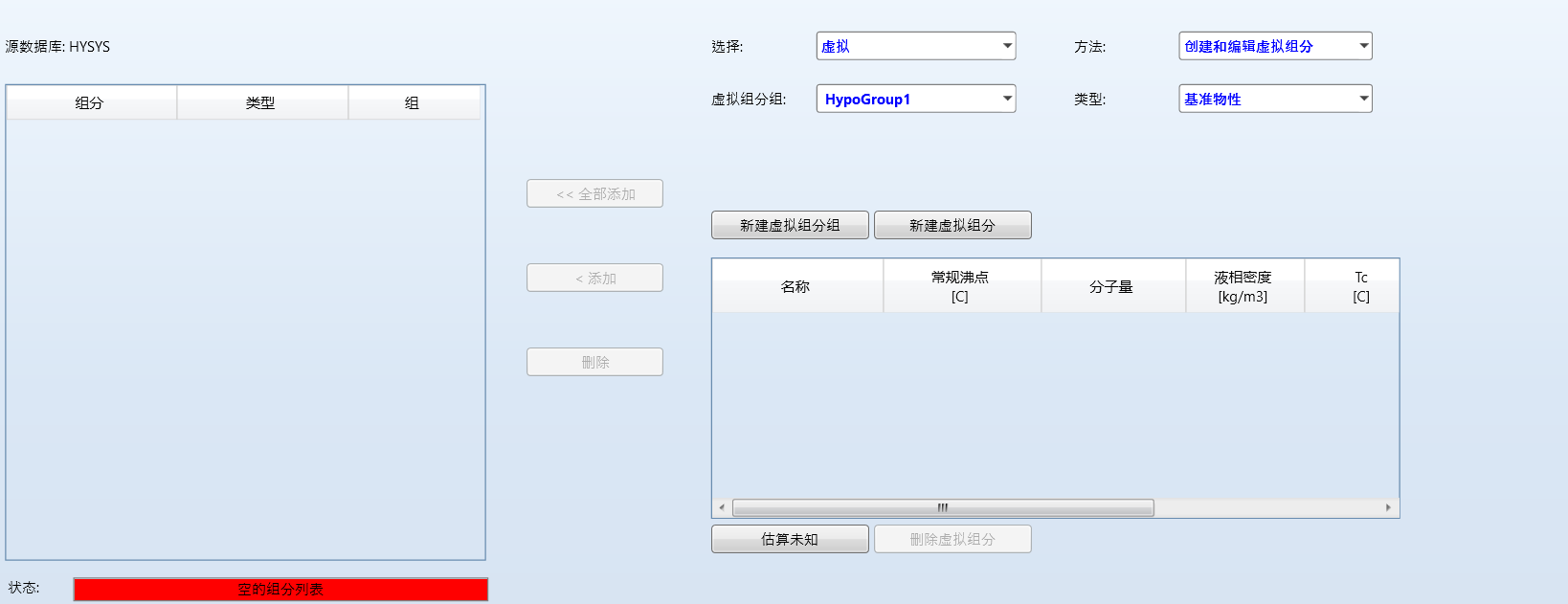
点击新建组分:
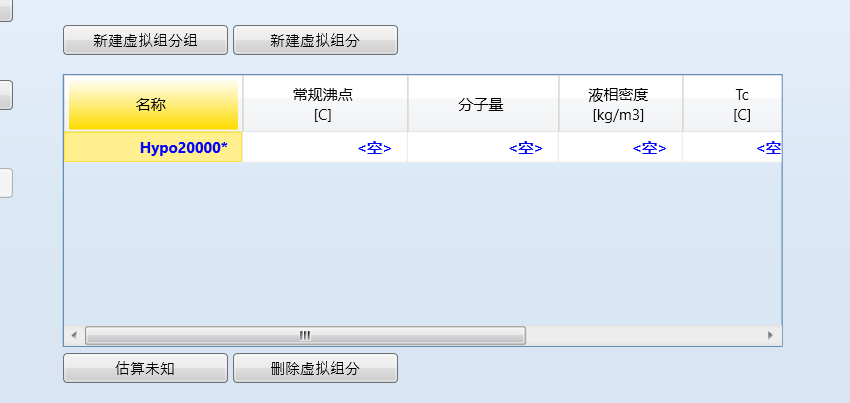
双击组分,进入编辑界面,输入组分名称:
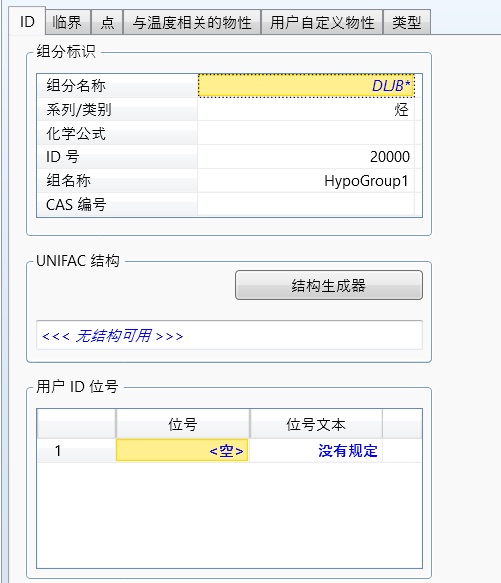
点击结构生成器,输入结构:
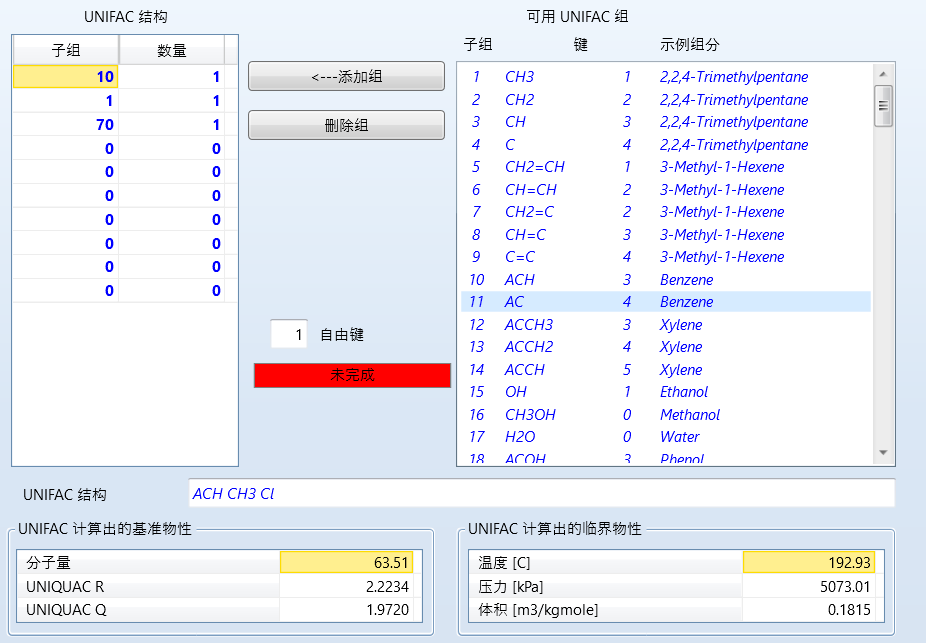
这里我只是举例,所以输入的不准确,一般来说自由键应该是刚刚好的才最准确
继续输入沸点和分子量:
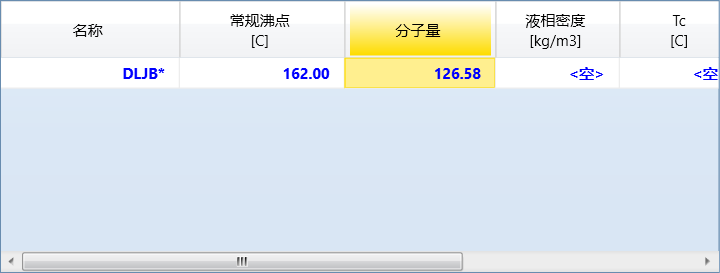
点击下方的估算未知,然后点击添加组分:
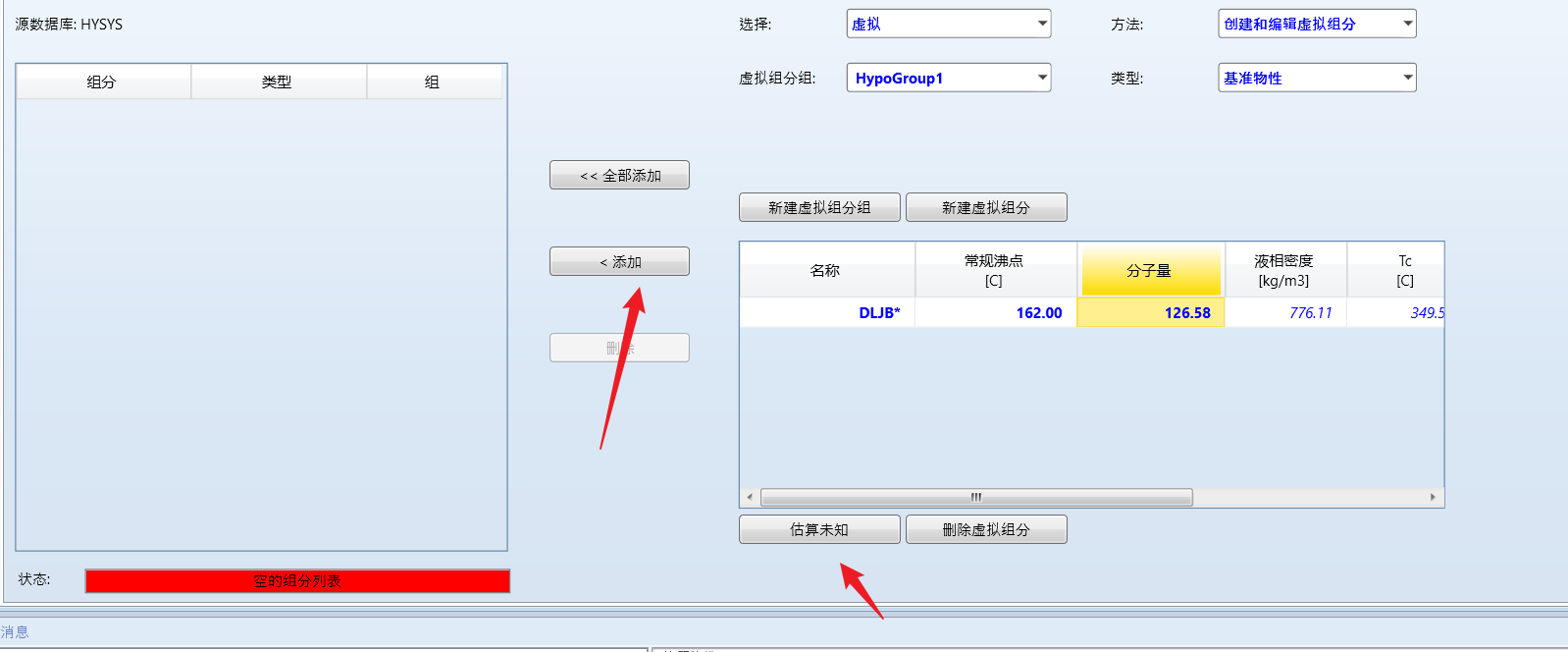
然后选择物性方法就可以进行模拟了
ProII
打开proii,新建文件,选择组分选择:
选择user-defined:
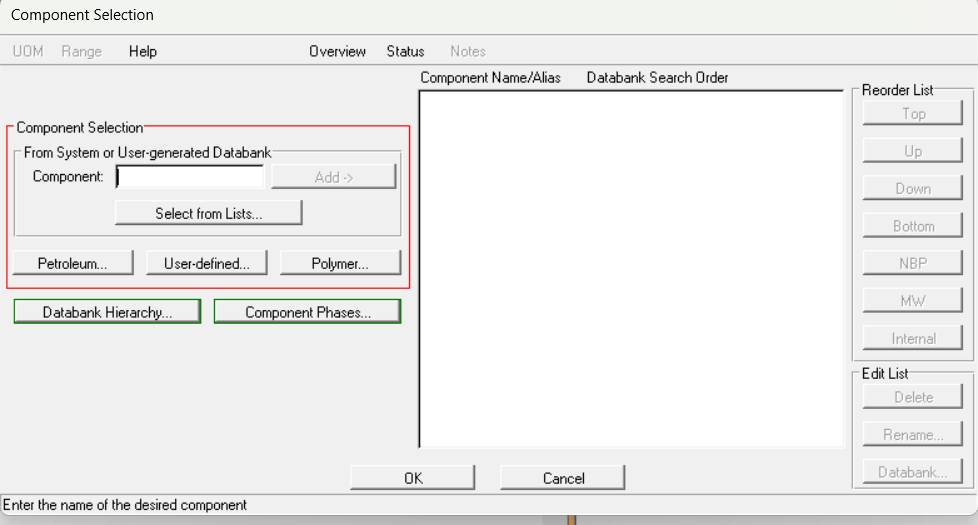
输入组分名称,点击add:
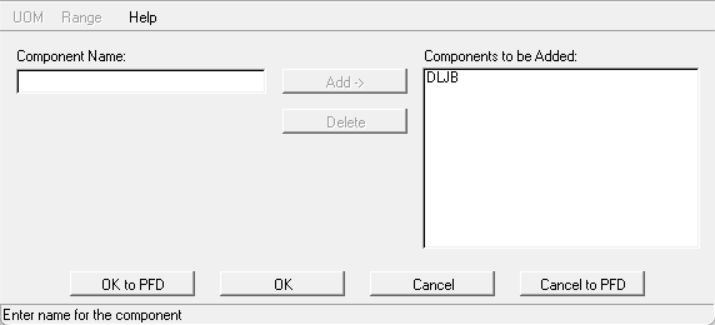
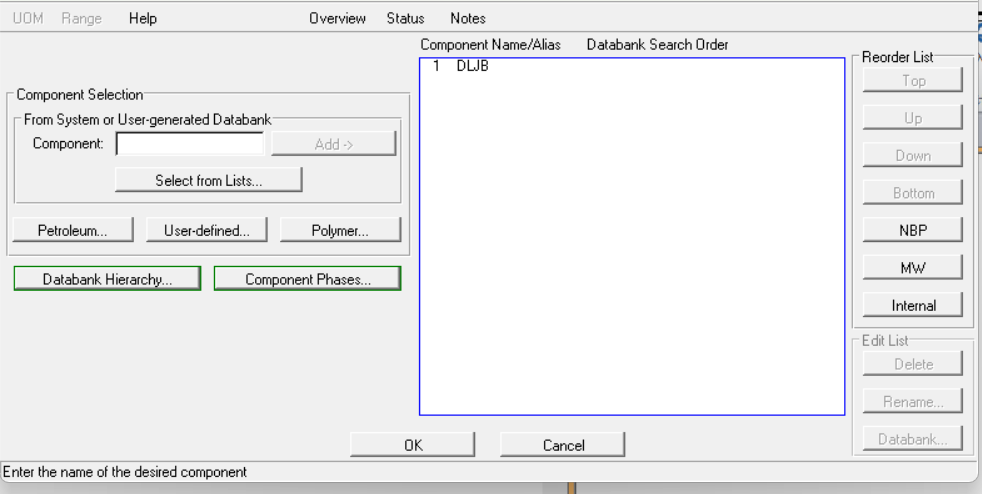
点击OK关闭即可
打开输入-组分性质页面
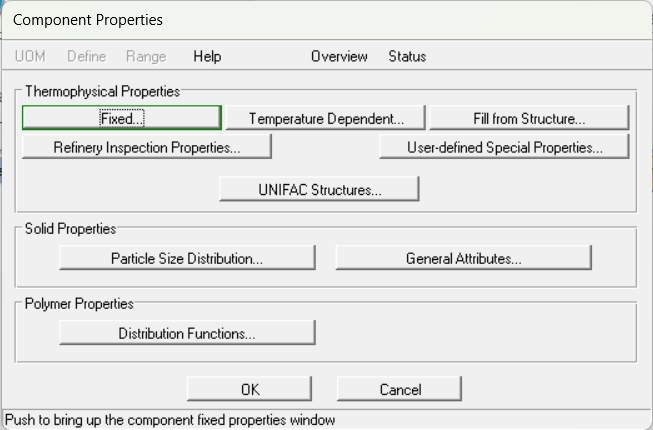
打开fixed页面,输入沸点和分子量:
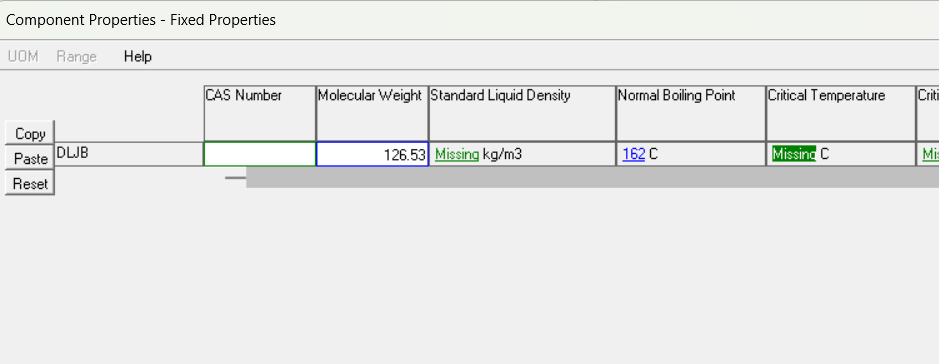
点击ok关闭,回到上一个界面,点击fill from structure,把自定义组分添加到估算列表里:
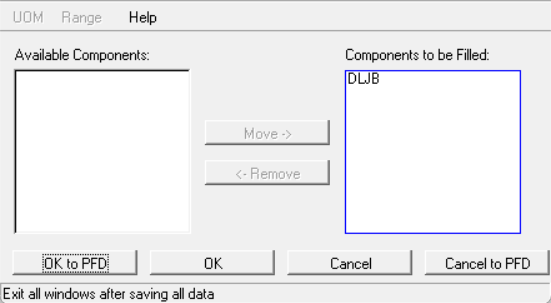
点击ok关闭,回到上一个页面,可以看到UNIFAC是红框,点击UNIFAC输入界面:
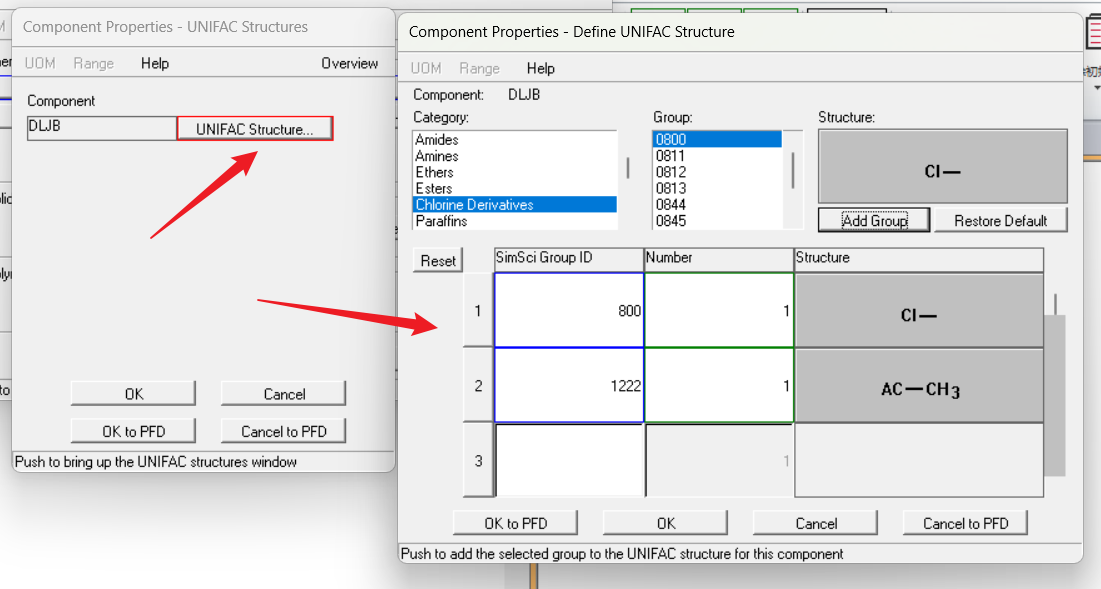
点击ok关闭,选择热力学方法即可开始模拟
总结
虽然今天举得例子不太恰当,但是也完美的演示了如何进行自定义组分的输入,但是这并不代表估算的性质就一定准确,后面会继续更新如何准确的估算物质性质,敬请期待,嘻嘻